What is GMP & Why & Who Needs To Comply?
What is GMP?
GMP refers to the Good Manufacturing Practice Regulations promulgated by the US Food and Drug Administration under the authority of the Federal Food, Drug, and Cosmetic Act.
GMP regulations address issues including record keeping, personnel qualifications, sanitation, cleanliness, equipment verification, process validation, and complaint handling. Most GMP requirements are very general and open-ended, allowing each manufacturer to decide individually how to best implement the necessary controls.
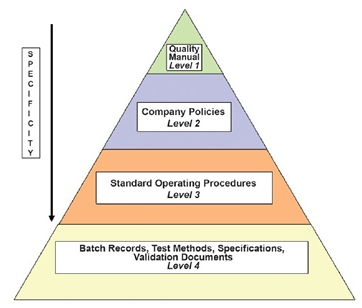
The organization should establish a hierarchical document system as shown below:
Who Needs To Comply With GMP & Why?
Who? GMP regulations require that manufacturers, processors, & packagers of drugs, medical devices, some food, & blood take proactive steps to ensure that their products are safe, pure, and effective. GMP regulations require a quality approach to manufacturing, enabling companies to minimize or eliminate instances of contamination, mixups, & errors. This in turn, protects the consumer from purchasing a product which is not effective or even dangerous.
Why? Failure of firms to comply with GMP regulations can result in very serious consequences including recall, seizure, fines, and jail time.
What are the various types of documents that a GMP facility need to maintain?
- Quality manual: A global company document that describes the regulations that the company is required to follow.
- Policies: Documents that describe in general terms how specific GMP aspects will be implemented.
- Standard operating procedures (SOPs): Step-by-step instructions for performing operational tasks or activities.
- Batch records: Documents maintained by the manufacturing department with step-by-step instructions for production-related tasks and activities
- Test methods: Documents completed by the QC department with step-by-step instructions for testing supplies, materials, products, and other production-related tasks and activities of the GMP facility.
- Specifications: Documents that list the requirements that a supply, material, or product must meet before being released for use or sale.
- Logbooks: Bound collection of forms used to document activities of operation, maintenance, calibration of a piece of equipment and record critical activities, change controls and its corrective action assignment.
How does LuitBiz DMS & QMS help you become GMP compliant?
Chapter
What it means
How LuitBiz DMS & QMS helps
6.10: Audit Trail
All documents related to the manufacture of intermediates or APIs should be prepared, reviewed, approved, and distributed according to written procedures. Such documents can be in paper or electronic form.
LuitBiz DMS & QMS provides full audit trail of all actions within the repository that can’t be accessed or modified by any user.
6.11: Version Control
The issuance, revision, superseding, and withdrawal of all documents should be controlled by maintaining revision histories.
LuitBiz DMS & QMS have a built-in version control system that helps manage versions and historical records of all document & process form versions.
6.12 & 6.13: Records Retention
A procedure should be established for retaining all appropriate documents and the retention periods for these documents should be specified.
LuitBiz DMS has a feature to define document retention period for different groups of documents and alerts for their deletion on completion of their retention period. Hence, all production, control, and distribution records can be retained for at least 1 year after the expiry date of the batch and for APIs with retest dates, records can be retained for at least 3 years after the batch is completely distributed and these can all be configured in LuitBiz DMS.
LuitBiz QMS has a feature allow you to distribute controlled copies of the approved documents during the entire life-cycle of the document
6.16: Document In Original Format
Specifications, instructions, procedures, and records can be retained either as originals or as true copies such as photocopies, microfilm, microfiche, or other accurate reproductions of the original records. Where reduction techniques such as microfilming or electronic records are used, suitable retrieval equipment and a means to produce a hard copy should be readily available.
LuitBiz DMS stores files in their original format and hard copies can be printed out whenever necessary. Documents can be arranged in the 4-level folder structure as specified by the GMP regulations and user access rights can be given to the documents at the folder level.
LuitBiz QMS allows you to write documents online and hard copies can be printed out whenever necessary. Documents can be arranged in the 4-level folder structure as specified by the GMP regulations and user access rights can be given to the documents at the folder level.
6.17: Specification Records
Specifications should be established and documented for raw materials, intermediates where necessary, APIs, and labeling and packaging materials. In addition, specifications may be appropriate for certain other materials, such as process aids, gaskets, or other materials used during the production of intermediates or APIs that could critically affect quality. Acceptance criteria should be established and documented for in-process controls.
LuitBiz DMS allows group leaders to define in-process controls in the form of document tagging to specify information fields that need to be collected for raw materials, intermediates, APIs, and labeling and packaging materials. These information can be filled out and attached to the documents. Document search can also be performed on the values of these fields which makes document retrieval very fast and easy.
LuitBiz QMS allows very fast and easy retrieval of documents based on their content and document types. Additionally, process forms in LuitBiz QMS can be retrieved easily with its powerful search features.
6.61: Change Control
Complete records should also be maintained for any modifications to an established analytical method, periodic calibration of laboratory instruments, apparatus, gauges, and recording devices, all stability testing performed on APIs and out-of-specification (OOS) investigations
LuitBiz DMS maintains all the change control records in the document's audit trail.
LuitBiz QMS allows users to request for change control, get their requests approved and maintains a complete audit trail of all the change control requests.